|
Основной целью СММ ("Сектора Молекулярного Моделирования") является компьютерное моделирование и анализ биофизических и радиобиологических экспериментов, проводимых в ОИЯИ. В качестве инструмента моделирования применяется стандартный и квантово-химический молекулярно-динамические методы для исследования структурных свойств биологических структур (ДНК, РНК и протеины) в растворах и различных средах. В этих целях имеются вычислительные мощности - кластеры рабочих станций, разработанные в СММ, а также компьютер специального назначения MDGRAPE-2, предоставленный нам РИКЕНом, Япония
Источники многих заболеваний связаны с изменениями, происходящими в белковых структурах клеток, причиной которых отчасти могут быть мутации – замещение одной или нескольких аминокислот в структуре белка. Экспериментальное изучение мутационных явлений в биологических белковых структурах традиционными рентгеноструктурными или ядерными магнитно-резонансными методами представляет собой исключительно трудоемкую и дорогостоящую задачу. Несмотря на современный прогресс экспериментальной техники, определение структурных и динамических свойств белков – очень трудоемкая задача, отнимающая немало времени. Поэтому решение этих важных задач в настоящее время под силу только компьютерному моделированию на базе молекулярной динамики. Вместе с тем, моделирование структуры белков необходимо для разработки препаратов, направленных на то, чтобы компенсировать отрицательный эффект мутаций, усилить или ослабить активность определенных генов.
Молекулярное моделирование является одним из важнейших инструментов для исследования био- и наноструктур, в биоинженерии и биодизайне, в процессе создания и оптимизации современных технологических установок (суперкомпьютеров), обладающих сложной пространственной конфигурацией и множеством определяющих параметров системы. Адекватная физико-математическая модель в таких задачах предполагает учет множества взаимосвязанных разномасштабных био-физических и физико-химических процессов. Компьютерное моделирование процессов столь высокой сложности предъявляет чрезвычайно высокие требования к эффективности применяемых методов и алгоритмов, а также к используемым вычислительным ресурсам. Проведение таких расчетов возможно только с использованием суперкомпьютеров или высокопроизводительных вычислительных кластеров.
Сотрудники сектора КММ (Компьютерного молекулярного моделирования) проводят научно-исследовательские и образовательные работы по следующим направлениям
Проект 1: «Молекулярная динамика хромофора родопсина - 11-цис ретиналя и окружающих амино кислотных остатков в хромофорном участке при физиологической регенерации зрительного пигмента: компьютерное моделирование»
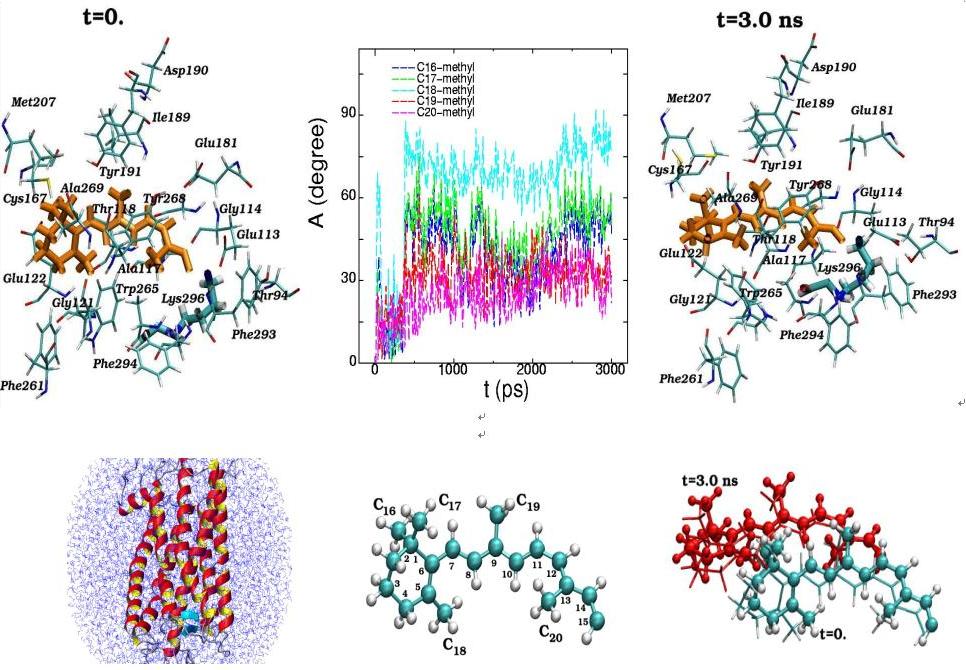
Методом компьютерного моделирования проведено сравнительное исследование молекулярной динамики родопсина, содержащего хромофорную группу (11-цис ретиналь), и свободного опсина. Молекулярная динамика прослежена во временном интервале, равном 3000 пикосекунд; при этом получено и проанализировано 3х106 дискретных конформационных состояний родопсина и опсина. Продемонстрировано, что «встраивание» хромофорной группы в хромофорный центр опсина оказывает существенное влияние на ближайшее белковое окружение хромофора, на конформационное состояние цитоплазматического домена и, практически, не оказывает влияния на конформационное состояние внутридискового домена. На основании результатов моделирования обсуждается возможный внутримолекулярный механизм поддержания родопсина как G-белок связывающего рецептора в неактивном состоянии, т.е. функция хромофора как эффективного лиганда-антагониста.
Molecular dynamics of 11-cis-retinal in the rhodopsin chromophore center at the initial (t=0) and final (t=3 ns) simulation states are presented along with the torsion rotation angles of five methyl groups (C16-C20) (top). The positions of the11-cis-retinal atoms during the 3 ns dynamical changes are separately displayed (bottom). (View from the side of the rhodopsin molecule)
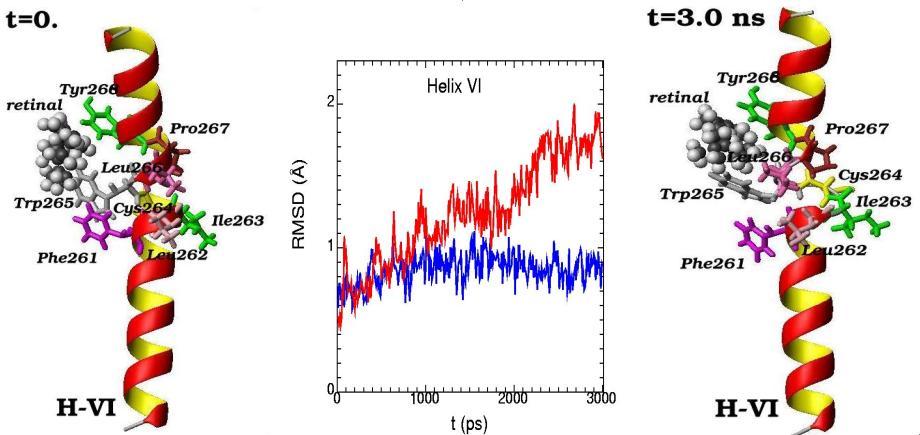
Molecular dynamics of alpha-helix H-VI and its amino acid residues surrounding the 11-cis-retinal chromophore are presented for the initial (t=0) and final (t=3 ns) simulation states. (The 11-cis-retinal is shown as balls and amino acid residues are shown as 3D structural formulas). The values of RMSD (root-mean-square-deviation) during the 3 ns molecular dynamics changes are shown for alpha-helix H-VI (middle picture). Blue curve – displays the deviation from the reference structure for free opsin (without retinal chromophore), red curve – for rhodopsin (with 11-cis retinal chromophore)
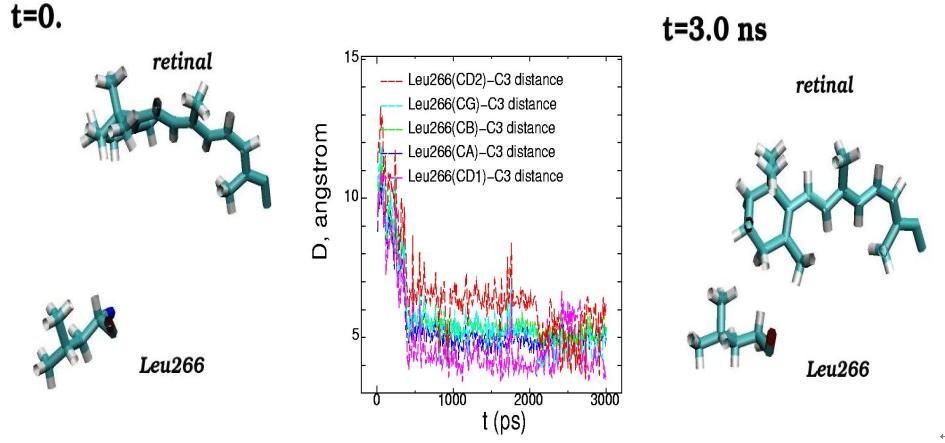
 Molecular dynamics of 11-cis-retinal chromophore and amino acid residue Leu266 of rhodopsin are presented for the initial (t=0) and final (t=3 ns) simulation states. (Side view from the rhodopsin cytoplasmic part). The plots of the interatomic distances between the different atoms of Leu266 and atom C3 of the β-ionone ring of 11-cis-retinal are displayed for the 3 ns molecular dynamics changes (middle picture)
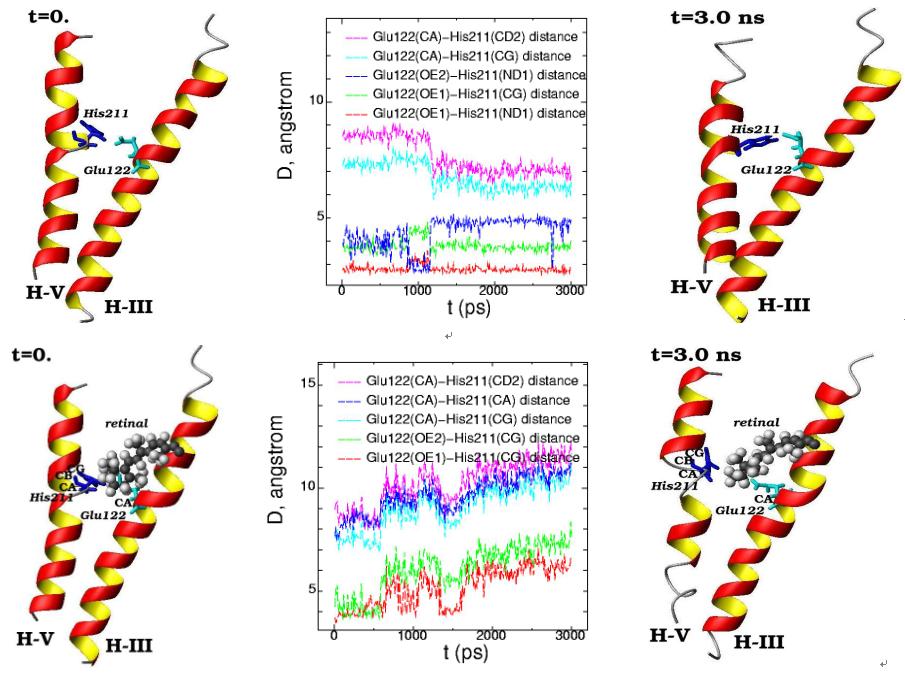
Molecular dynamics of the alpha-helices H-III (Glu122) and H-V (His211) for the initial (t=0) and final (t=3 ns) simulation states are presented in free opsin (rhodopsin without retinal chromophore) (top) and in rhodopsin (with 11-cis-retinal chromophore) (bottom). (11-cis-retinal is shown as balls and amino acids are shown as 3D structural formulas). The plot of the interatomic distances between the different atoms in Glu122 and His211 are displayed for the 3 ns molecular dynamics changes (middle pictures)
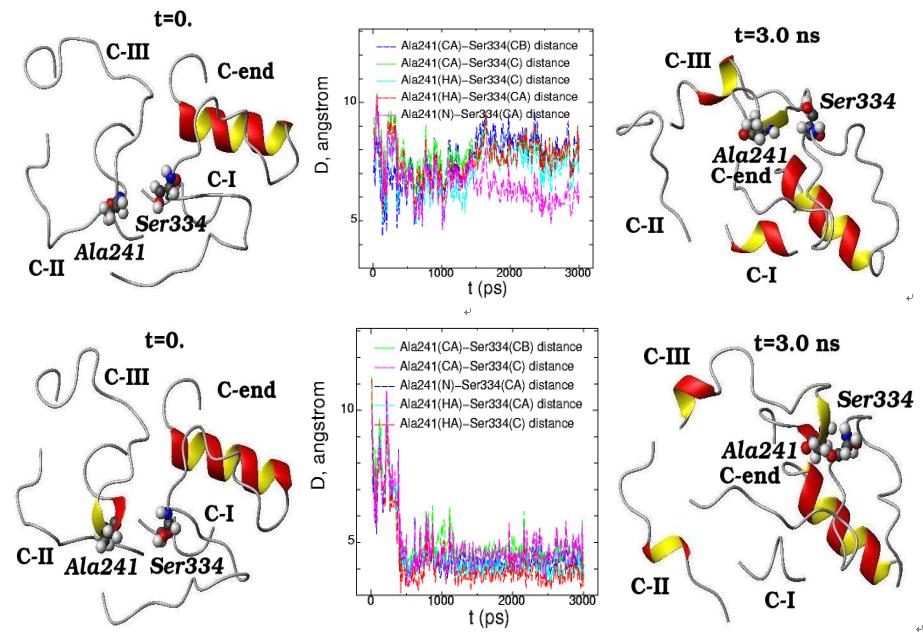
Molecular dynamics of C-end peptide (Ser334) and C-II loop (Ala241) for the initial (t=0) and final (t=3 ns) simulation states are presented in free opsin (rhodopsin without retinal chromophore) (top) and in rhodopsin (with 11-cis-retinal chromophore group) (bottom). The plot of interatomic distances between the different atoms in Ser334 and Ala241 are displayed for the 3 ns molecular dynamics changes (middle pictures)
Проект 2: «МД анализ связей АТФ, субстрата и белковых компонентов киназы с помощью МД моделирования кристаллической решетки двухкомпонентной активной CDK2»
В рамках настоящего проекта проведены широкомасштабные молекулярно-динамические (МД) моделирования циклин-зависимых протеинкиназ (CDK) с комплексом АТФ. Протеинкиназа CDC28 дрожжей Saccharomyces cerevisiae служит привлекательной моделью для исследования механизмов регуляции киназ, актуальность изучения которых обусловлена центральной ролью протеинкиназ CDK в регуляции клеточного цикла и высокой частотой нарушения CDK или дерегуляции ингибиторов CDK при злокачественном перерождении клеток млекопитающих. Для анализа структурных изменений, к которым приводит замена СDС28-G20S, использовали кристаллическую структуру киназы человека CDK2. По данным МД-моделирования структура немутантного и мутантного, включающего замену G16S-CDK2, соответствующую дрожжевой G20S-СDС28, комплексов CDK2 заметно отличаются друг от друга, причем различия структурных конформаций наиболее ярко проявляются именно в тех участках (например, G- и T-петли), которые играют ключевую роль для функционирования киназы.
Центральная роль, которую циклин-звависимые киназы играют на протяжении всего цикла деления клетки и большое влияние генетических изменений CDK или дерегуляции CDK ингибиторов на число раковых клеток, делают CDC28 дрожжей Saccharomyces cerevisiae чрезвычайно привлекательной моделью для изучения механизма регуляции CDK. Нами было обнаружено, что определенные генные мутации, включающие cdc28-srm, влияют на ход цикла клетки, содержание различных генетических структур и повышают чувствительность клеток к ионизирующему излучению. Мутация cdc28-srm не является температурно-чувствительной и отличается от известной cdc28-ts так как последняя имеет фенотипические черты при 30oC. Последовательный анализ cdc28-srm показал одиночное нуклеотидное замещение G20S. Это является третьим глицином в сохраняющейся последовательности GxGxxG в G-обогащенной петле, находящейся напротив активационной T-петли. Несмотря на всю продемонстрированную важность, роль G-петли остается неясной. Кристаллическая структура человеческой CDK2 послужила моделью каталитического остова других CDK, включая CDC28. Были проанализированы молекулярно-динамические (МД) траектории CDK2/ATP комплекса наносекундной длительности. МД исследования замещения CDK2-G16S (CDC28-G20S) показали конформационные отличия результирующей структуры CDK2 в отдалении G-петли от ATP и новое перераспределение аминокислот в T-петле.
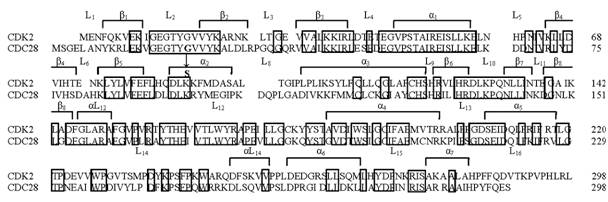
Выравненные N-концевые фрагменты последовательности циклинзависимых киназ человека (CDK2) и дрожжей S. cerevisiae (CDC28). Рамкой обведены остатки, консервативные для четырех киназ - человека (CDK2, CDC2) и дрожжей (cdc2, CDC28). Отмечены элементы вторичной структуры – линкерные области (L) между структурными элементами (β-нити, α-спирали).
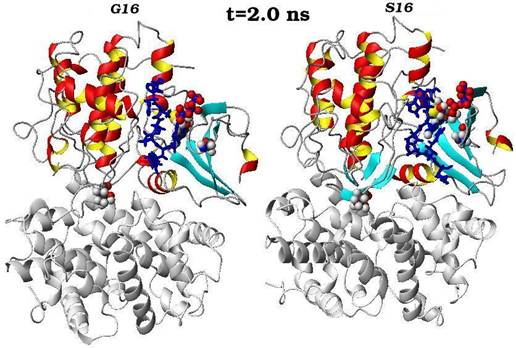
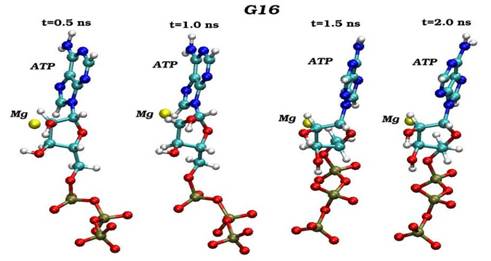
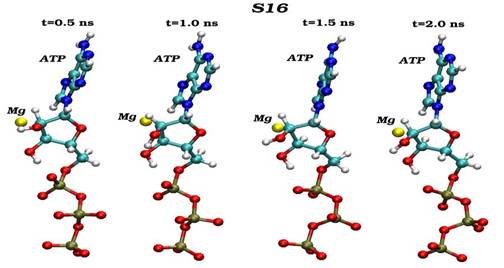
Конечные структурные конформации по данным молекулярно-динамических расчетов нативного (G16) и мутантного (S16) комплексов CDK2. Конформации белковых структур построены на основе расчета равновесных положений атомов комплексов СDК2 в интервале времени 2 нс, в качестве начальной структуры использовалась структура кристаллической решетки, полученной с помощью рентгеноструктурного анализа.
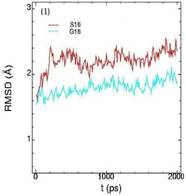
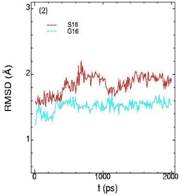
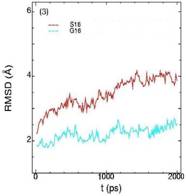
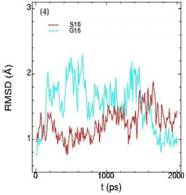
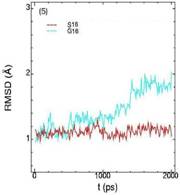
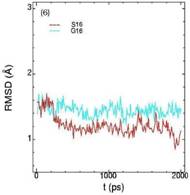
Значения среднеквадратичных отклонений смещений атомов (RMSD) различных частей белкового комплекса активной киназы CDK2: (1) киназа (ост. 1-296); (2) циклин (ост. 297-554); (3) суммарно киназа и циклин; (4) субстрат (ост. 557-563); (5) T-петля (ост. 147-167) и (6) G-петля (ост. 11-18). Нумерация аминокислотных остатков идет в соответствии с нумерацией атомов в структуре исходного файла базы данных PDB.
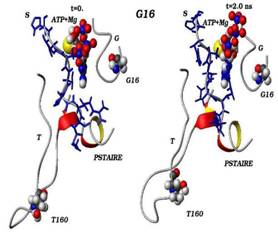
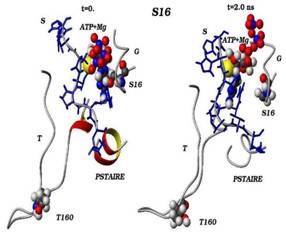
Сравнения начальных (t=0) и конечных (t=2 нс) конформаций для «ключевых структурных элементов» нативного (G16) и мутантного (S16) комплексов CDK2. Указаны сайт мутационного замещения G/S16 и сайт фосфорилирования T160, комплексы АТФ-Mg2+, фрагмент PSTAIRE, а также позиций G-, T-петель и субстрата S.
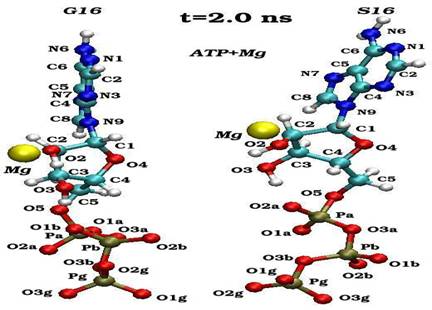
Пространственные ориентации молекулы АТФ по отношению к иону магния в конечном состоянии (2 нс) для нативного (G16) и мутантного (S16) комплексов
Динамика пространственных ориентаций молекулы АТФ по отношению к иону магния для нативного (G16-верхний снапшот) и мутантного (S16-нижный снапшот) комплексов.
Проект 3: «МД моделирование механизмов SOS мутагенеза в Escherichia coli на основе изучения конформационного поведения белков LexA и RecA, участвующих в формировании связей двух-нитевых структур ДНК»
В рамках данного проекта нами были выполнены гомологичные и молекулярно-динамические (МД) моделирования белка RecA с целью изучения влияния аминокислотных мутационных замещений в бета-листовых и ингибирующих по отношению к ДНК сайтах белка. RecA белок представляет собой один из важных ферментов, участвующих в механизмах SOS репарации и восстановлении структуры ДНК при различных повреждениях ДНК. Мономерная структура белка RecA содержит в своем центральном домене два связывающие по отношению к ДНК сайта: один - для связывания одно-нитевой ДНК (ssDNA), другой- для двух-нитевой ДНК (dsDNA); при этом эти сайты связывания ДНК включают в себя также две неупорядоченные петли (L1 & L2). Как показывают эксперименты, указанные петли представляют собой существенно важные структурные элементы - мутации в этих неупорядоченных областях могут привести к ингибированию сайта связывания ДНК (например, точечные мутации могут быть в L2: Gly204, Glu207, и Gly211; в L1: Gly160, Gly157, и Arg169) . В данной работе нами проводится оценка влияния мутационного замещения Gly204Ser на динамические и структурные свойства RecA. Основываясь на гомологичном моделировании, мы предсказали неизвестные конформации вышеуказанных неупорядоченных петель L1 & L2, затем провели МД вычисления над целой восстановленной структурой RecA белка. Сравнения динамических и структурных свойств белков дикого (нативного) и мутантного типа нами прослеживалась при одинаковых условиях моделирования до 2-нс конформационного состояния.
Проект 4: «Образовательная программа по направлению курса молекулярной динамики в исследовании био- и наноструктур»
Будет продолжена работа со студентами и аспирантами по изучению и применению курса компьютерного МД-моделирования для радиобиологических и биофизических задач по теме сектора, лаборатории и института
|