|
The main objective of the CMM ("Center for Molecular Modeling") is computer simulation and analysis to support the biophysical and radiobiological experiments performed at JINR. As for simulation tools we employ conventional and ab initio molecular dynamics methods to investigate structural properties of biophysical systems (DNA,proteins,etc.) in water and solvent environment. For the computer tools we use a cluster of workstations, developed at CMM and a special-purposes MDGRAPE-2 computer for MD simulations, which is kindly supported by the Computational Astrophysics Laboratory of RIKEN.
Computer molecular simulations of complex multiparticle systems play a fascinating role in fundamental physics, biochemical and life sciences. Having an increasingly significant impact on many applied industries, especially in modern biophysical and nanotechnological areas, molecular simulation provides a set of tools for predicting many functional properties of molecular systems. The chemical, pharmaceutical, materials and related industries - all share the computer molecular simulation methods. The molecular simulation studies cover different fields of either biological processes - protein folding and electron densities of DNA and proteins, or thin film formations and surface-cluster phenomena in nanoelectronics, synthetic copolymers and biopolymer design in biochemistry, so on. Practically all of the world's present supercomputers and many specially developed high performance computing clusters over the world are performing molecular simulations or are aimed on these needs.
The origin of many diseases in human body has to correlate with the changes in the protein function followed by their improper conformational behavior and unfolding. The cause of such changes might be the substitution of even one or few specific amino acid residues in protein structure. The experimental studies of mutation transitions for protein structures by the traditional X-ray or NMR measurement represent themselves as the extremely difficult, expensive and time-consuming task. Using the adequate computational methods (molecular dynamics), based on their efficient implementation in the parallel/vector and special-purpose (MDGRAPE-2 and 3) machines, would allow ones to simulate the valid behavior of the wild-type and mutated proteins in water or ionic solvents at reliable physiological temperatures and conditions. Thus, the molecular simulation studies between the wild-type and mutant proteins represent themselves as a fundamental problem of great importance. The correlation of the conformational behavior between the wild-type and mutant proteins (RecA and MutS proteins, cyclin-dependent kinases and visual pigment rhodopsin) have been investigated and answered in the present study.
The staff of CMM sector are performing the scientific research and educational activity within the following topics (projects planned by LRB):
Project 1: «Molecular Dynamics of Chromophore 11-cis Retinal And Surrounding Amino Acid Residues In The Chromophore Binding Pocket at Physiological Regeneration of Visual Pigment Rhodopsin: Computer Simulation»
Molecular dynamics simulations are carried out on the rhodopsin protein to investigate the conformational changes of the protein in relation to the inclusion of the 11-cis chromophore retinal into consideration. Molecular dynamics calculations were performed for the time interval from t=0 to 3000 picoseconds, so that the configuration states of rhodopsin and free opsin were analyzed and compared. It was demonstrated that the adaptation of the chromophore retinal in the opsin site causes a considerable influence on its protein binding pocket, as well as on conformations of the cytoplasmic part, but the extracellular part of the protein shows a comparably small changes. On the basis of the simulation results we discuss some molecular mechanisms for the rhodopsin protein function as a G-protein-coupled receptor in the dark state, i.e. for the chromophore retinal as a ligand-agonist stabilizing the inactive conformation of the rhodopsin.
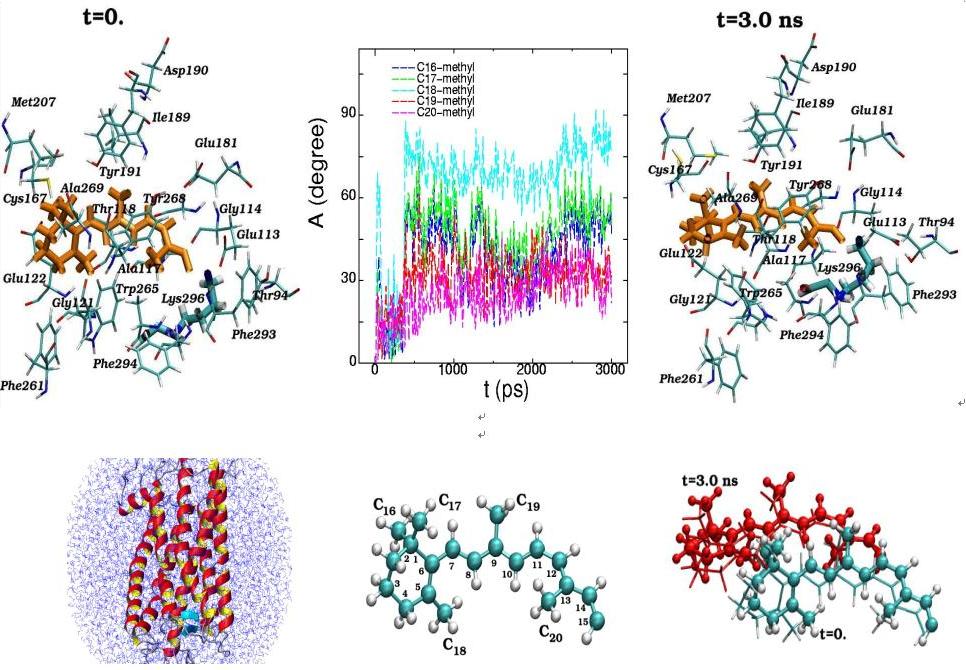
Molecular dynamics of 11-cis-retinal in the rhodopsin chromophore center at the initial (t=0) and final (t=3 ns) simulation states are presented along with the torsion rotation angles of five methyl groups (C16-C20) (top). The positions of the11-cis-retinal atoms during the 3 ns dynamical changes are separately displayed (bottom). (View from the side of the rhodopsin molecule)
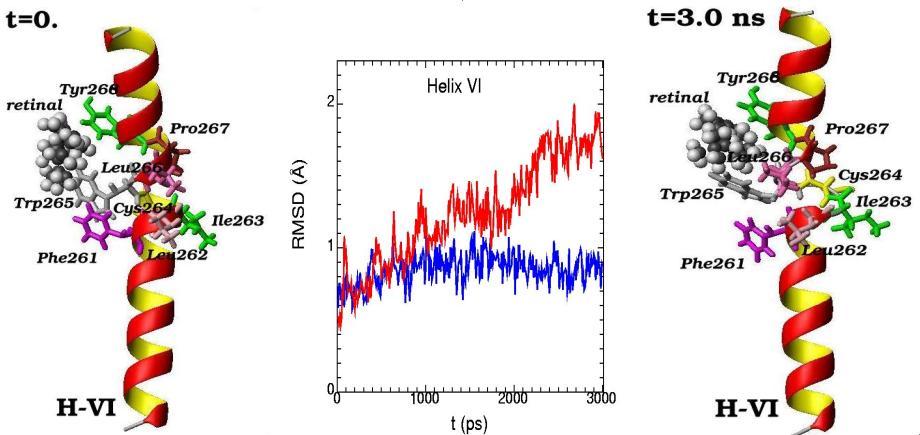
Molecular dynamics of alpha-helix H-VI and its amino acid residues surrounding the 11-cis-retinal chromophore are presented for the initial (t=0) and final (t=3 ns) simulation states. (The 11-cis-retinal is shown as balls and amino acid residues are shown as 3D structural formulas). The values of RMSD (root-mean-square-deviation) during the 3 ns molecular dynamics changes are shown for alpha-helix H-VI (middle picture). Blue curve – displays the deviation from the reference structure for free opsin (without retinal chromophore), red curve – for rhodopsin (with 11-cis retinal chromophore)
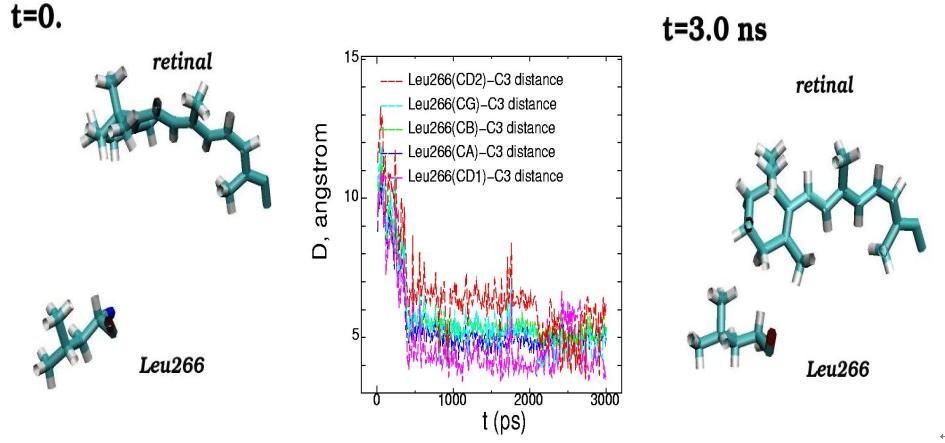
 Molecular dynamics of 11-cis-retinal chromophore and amino acid residue Leu266 of rhodopsin are presented for the initial (t=0) and final (t=3 ns) simulation states. (Side view from the rhodopsin cytoplasmic part). The plots of the interatomic distances between the different atoms of Leu266 and atom C3 of the ?-ionone ring of 11-cis-retinal are displayed for the 3 ns molecular dynamics changes (middle picture)
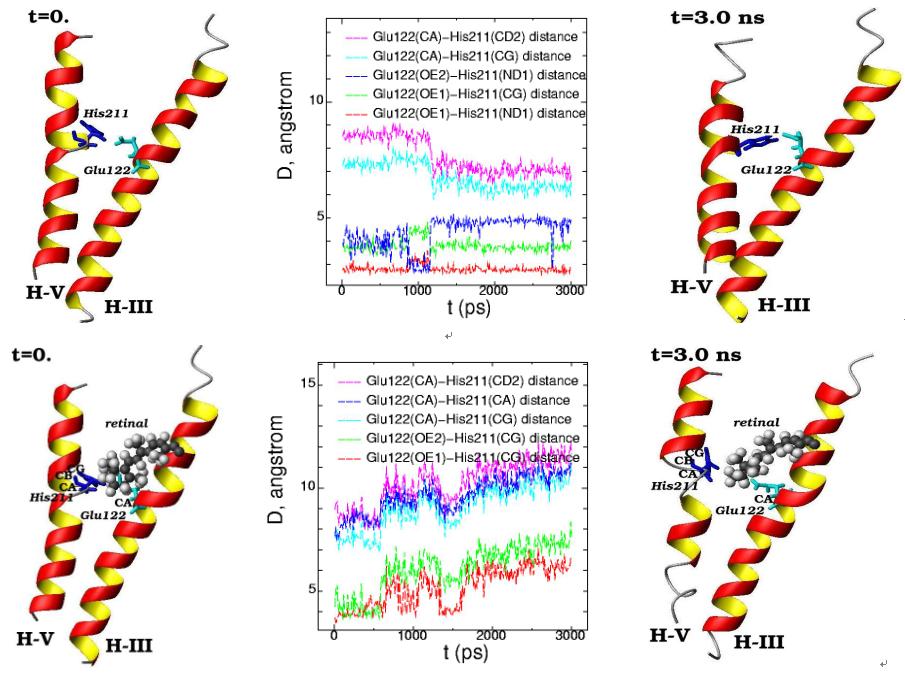
Molecular dynamics of the alpha-helices H-III (Glu122) and H-V (His211) for the initial (t=0) and final (t=3 ns) simulation states are presented in free opsin (rhodopsin without retinal chromophore) (top) and in rhodopsin (with 11-cis-retinal chromophore) (bottom). (11-cis-retinal is shown as balls and amino acids are shown as 3D structural formulas). The plot of the interatomic distances between the different atoms in Glu122 and His211 are displayed for the 3 ns molecular dynamics changes (middle pictures)
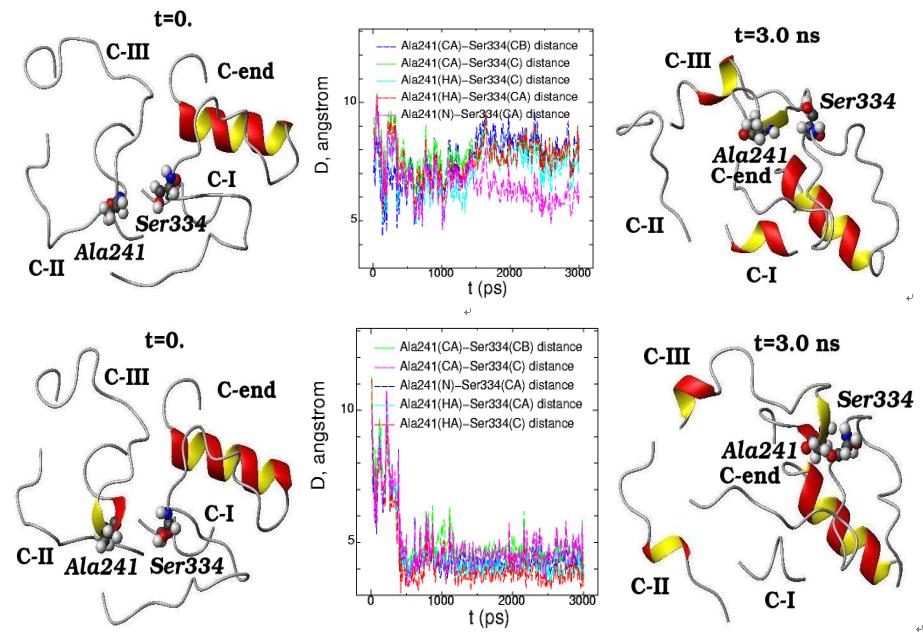
Molecular dynamics of C-end peptide (Ser334) and C-II loop (Ala241) for the initial (t=0) and final (t=3 ns) simulation states are presented in free opsin (rhodopsin without retinal chromophore) (top) and in rhodopsin (with 11-cis-retinal chromophore group) (bottom). The plot of interatomic distances between the different atoms in Ser334 and Ala241 are displayed for the 3 ns molecular dynamics changes (middle pictures)
Project 2: «MD Analysis of the Bonds Between the ATP and Catalytic Subunit of Kinases (Wild And Kinases) Using MD Simulations of The Active CDK2 Crystal Lattice»
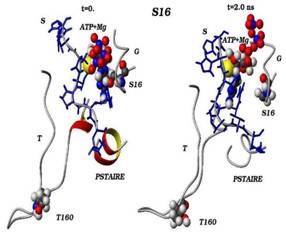
In the present work nanoseconds long molecular dynamics (MD) simulations of the cyclin-dependent proteinkinases (CDK) with ATP complex were performed. The central role that CDK play in the timing of cell division and repair and the high incidence of genetic alteration or deregulation of CDK inhibitors in a number of cancers make CDC28 of yeast Saccharomyces cerevisiae very attractive model for studies of mechanisms of CDK regulation. The crystal structure of the human CDK2 has served as a model for the catalytic core of other CDKs, including CDC28. MD simulations of substitution CDK2-G16S in conserved G-loop shows an important of this amino acid and a conformational change of CDK2 structure resulting in the moving of the G-loop away from ATP and a new rearrangement of amino acids in the T-loop.
The cyclin-dependent kinases play an essential role in the timing of cell division and repair, furthermore an high incidence of genetic alterations of CDKs or deregulation of CDK inhibitors have been observed in several cancers. These arguments make of CDC28 of Saccharomyces cerevisiae yeast a very attractive model to study CDK regulation mechanisms. We observed certain gene mutations, including cdc28-srm [G20S], affect cell cycle progression, maintenance of different genetic structures, checkpoint-control and increase cell radiosensitivity. A cdc28-srm mutation is not a temperature-sensitive mutation and differs from known cdc28-ts mutations, since it shows the evident phenotypic manifestations at 30oC. The mutation is on the third glycine site in the conserved sequence GxGxxG of the G-rich loop, whose position is opposite to the activation T-loop. Despite its established importance, the role of the G-loop is still unclear. The crystal structure of the human CDK2 served as model for the catalytic core of other CDKs, including CDC28. Nanoseconds long molecular dynamics trajectories of the CDK2/ATP complex were analyzed. The MD simulations of corresponded substitution CDK2-G16S in conserved G-loop shows this amino acid importance and the induced conformational change in CDK2 structure, resulting in the ATP removal from G-loop and in new amino acids rearrangement in the T-loop.
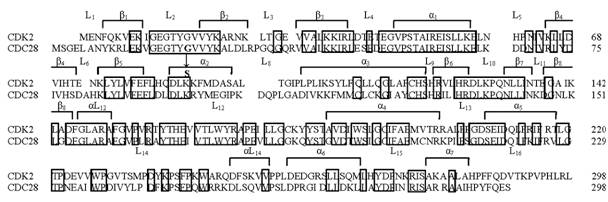
Выравненные N-концевые фрагменты последовательности циклинзависимых киназ человека (CDK2) и дрожжей S. cerevisiae (CDC28). Рамкой обведены остатки, консервативные для четырех киназ - человека (CDK2, CDC2) и дрожжей (cdc2, CDC28). Отмечены элементы вторичной структуры – линкерные области (L) между структурными элементами (β-нити, α-спирали).
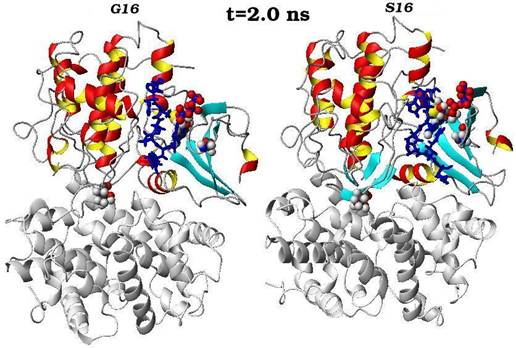
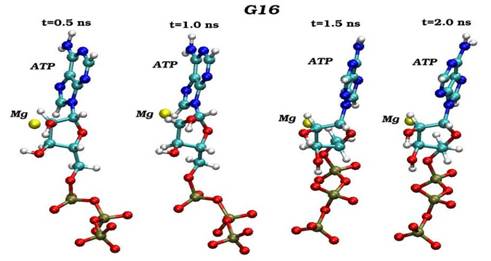
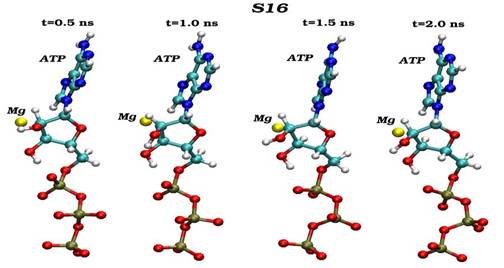
Конечные структурные конформации по данным молекулярно-динамических расчетов нативного (G16) и мутантного (S16) комплексов CDK2. Конформации белковых структур построены на основе расчета равновесных положений атомов комплексов СDК2 в интервале времени 2 нс, в качестве начальной структуры использовалась структура кристаллической решетки, полученной с помощью рентгеноструктурного анализа.
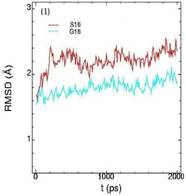
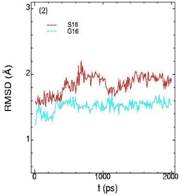
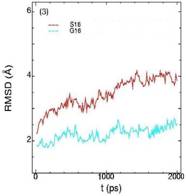
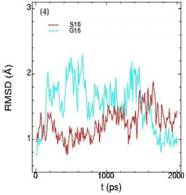
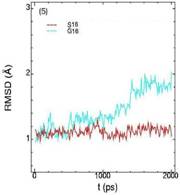
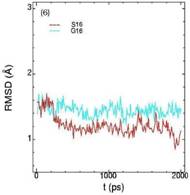
Значения среднеквадратичных отклонений смещений атомов (RMSD) различных частей белкового комплекса активной киназы CDK2: (1) киназа (ост. 1-296); (2) циклин (ост. 297-554); (3) суммарно киназа и циклин; (4) субстрат (ост. 557-563); (5) T-петля (ост. 147-167) и (6) G-петля (ост. 11-18). Нумерация аминокислотных остатков идет в соответствии с нумерацией атомов в структуре исходного файла базы данных PDB.
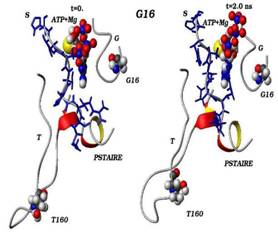
Сравнения начальных (t=0) и конечных (t=2 нс) конформаций для «ключевых структурных элементов» нативного (G16) и мутантного (S16) комплексов CDK2. Указаны сайт мутационного замещения G/S16 и сайт фосфорилирования T160, комплексы АТФ-Mg2+, фрагмент PSTAIRE, а также позиций G-, T-петель и субстрата S.
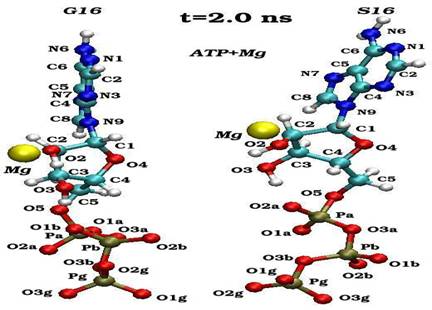
Пространственные ориентации молекулы АТФ по отношению к иону магния в конечном состоянии (2 нс) для нативного (G16) и мутантного (S16) комплексов
Динамика пространственных ориентаций молекулы АТФ по отношению к иону магния для нативного (G16-верхний снапшот) и мутантного (S16-нижный снапшот) комплексов.
Project 3: «MD Simulations of Mechanism of SOS Mutagenesis In Escherichia coli, Based On The Studies of The Conformation Bahevior of LexA and RecA Proteins Involved In The Formation of Single- And Double-Stranded DNA Structure»
Within the project we have performed homology modeling and molecular dynamics (MD) simulations on the RecA protein to study the influence of amino acid mutations in the beta-sheet and DNA inhibition sites. The RecA protein is one of the critical enzymes involved in SOS mechanism and stimulating DNA repair upon injury of DNA. The RecA monomer contains two DNA binding sites in its central domain, one for binding ssDNA, and another for binding duplex DNA, and both the DNA binding regions include disordered loops (L1 & L2). As experiments confirmed, the importance of the disordered regions in DNA binding and mutations in several residues in the disordered loops leading to inhibition of DNA binding (for example these residues are, in L2: Gly204, Glu207, and Gly211; in L1: Gly160, Gly157, and Arg169). In the present study we have estimated the effect of Gly204Ser mutation on the dynamical behaviour of RecA. Based on the homology modeling approach we have first predicted the conformation of above disordered loops L1 & L2, the next we have performed the MD calculations on the full RecA structure. The comparison analysis have been fulfilled between the wild-type and mutant RecA proteins within of 2-ns dynamical conformations and structural changes
Project 4: «Educational Programme On Teaching of The Molecular Dynamics Course In Studies of Bio- And Nano-Structures»
The results of the research work by the staff of the CMM sector are presented in All-Russian and International Conferences, Simposiums and Seminars, as well as published in the domestic and foreign journals (attached is Publication List).
|